A hypophysisdaganatok igen gyakran előforduló tumorok, kialakulásuk a morbiditás jelentős növekedésével jár. A familiáris hypophysisdaganatok az összes hypophysisadenomák mintegy 5%-át teszik ki. Előfordulhatnak öröklődő daganatszindrómák részjelenségeként, valamint úgy is, hogy az agyalapi mirigy adenomája mellett nem jelentkezik más jellegzetes tumor, ez esetben beszélünk familiáris izolált hypophysisadenomáról. A betegség öröklődő jellegére való tekintettel a családtagok genetikai és klinikai szűrővizsgálata az esetleges tumor jelenlétének mielőbbi felismerését és kezelését teszi lehetővé.
A hypophysisdaganatok gyakori intracranialis tumorok, előfordulási gyakoriságuk mintegy 1/1000 fő (1). Bár e daganatok általában jóindulatúak, a hormontúlprodukció és a nyomási tünetek számos súlyos szövődményt okozhatnak.
Első beteg
A 81 éves nőbeteget 2007 óta rendszeresen ellenőriztük ambulanciánkon hypophysismacroadenoma (27×17 mm) okozta acromegalia miatt. A beteg életkora, az előrehaladott vascularis encephalopathia, paroxysmalis pitvarfibrilláció és a súlyos szívelégtelenség miatt nem merülhetett fel a hypophysisműtét lehetősége.
Elsőként dopaminagonista-kezelést alkalmaztunk. A kontrollvizsgálatnál az orális glükóztolerancia-teszt (OGTT) során a beteg növekedésihormon- (GH-) szintje nem csökkent a megfelelő mértékre, ezért szomatosztatin (SS) -analóg-kezelésre tértünk át. Az SS-analóg-kezelés maximális dózisa mellett ugyan a daganat mérete csökkent (21,3×11,2×18,8 mm), de a napi profil során mért emelkedett GH-szintek alapján acromegaliája továbbra is aktív volt. Mindezek alapján további kezelésként radioterápia mellett döntöttünk. A kezelés hatásának kifejlődéséig folytattuk az SS-analóg-terápiát.
A 2011-es kontrollvizsgálatok során bár a daganat további csökkenését észleltük (17,9×10,8×15,9 mm), de biokémiai kontrollt továbbra sem sikerült elérni, így kezelését további gyógyszerrel, a GH-receptor-antagonista pegvisomant adásával egészítettük ki. Több kontrollvizsgálat nem történt, mivel a beteg meghalt.
Második beteg
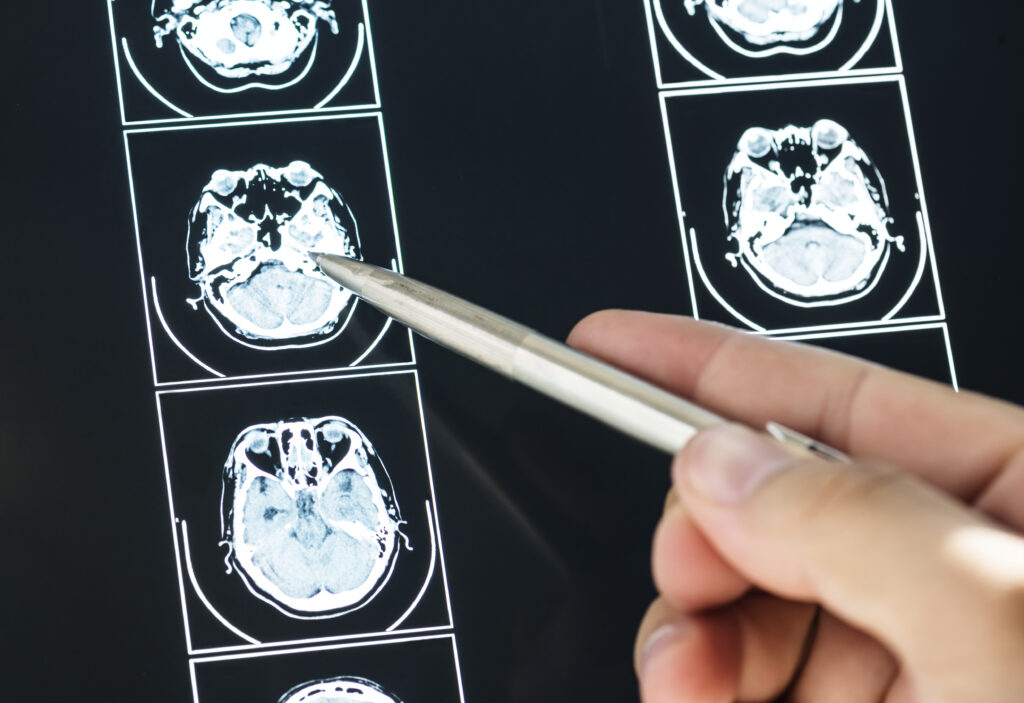
Az 54 éves férfi beteg 2008-ban került felvételre osztályunkra. Jelentősen emelkedett prolaktin- (PRL-), GH-szintekkel járó para- és suprasellarisan terjedő, 6 cm legnagyobb átmérőjű hypophysismacroadenomát (1. ábra) és következményes acromegaliát diagnosztizáltunk.

A dopaminagonista-kezelés megkezdését követő ellenőrzés alapján nem sikerült biokémiai kontrollt elérni, így terápiáját SS-analóggal egészítettük ki, majd 2009-ben műtétre került sor. A műtétet követően emelkedett hormonszinteket észleltünk, így tovább folytattuk gyógyszeres kezelését, kezdetben dopaminagonistával, majd ennek hatástalansága miatt SS-analóggal. A kontroll-MR-vizsgálat jelentős méretű residuumot igazolt. Ezt követően radioterápia mellett döntöttünk, a sugárkezelés hatásának kifejlődéséig tovább folytattuk gyógyszeres kezelését.
A 2012-ben elvégzett ellenőrző MR-vizsgálat azonban a tumor méretében progressziót mutatott, és a biokémiai kontroll sem volt kielégítő, ezért az SS-analóg-kezelés mellé a dopaminagonisták közül az acromegalia kezelésére a leghatékonyabbnak tartott szelektív hatású dopaminagonista – egyedi import alapján igényelhető – cabergolin adására tértünk át.
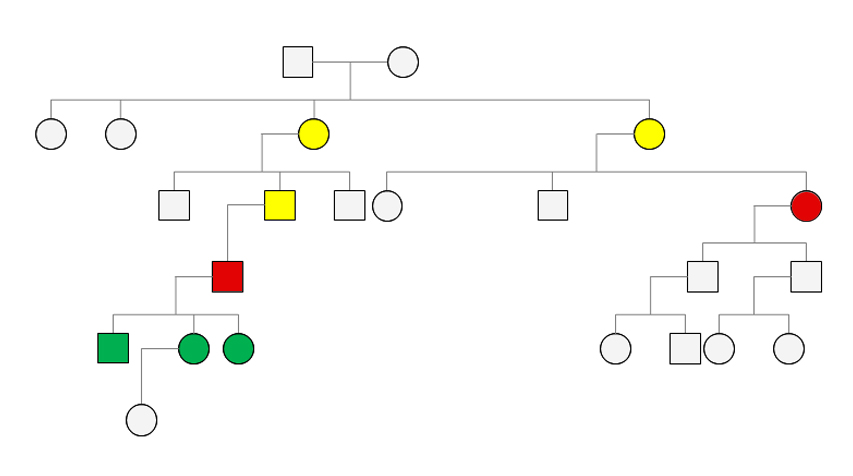
A férfi beteg anamnézisfelvételénél derült fény arra, hogy ugyanabból a faluból származik, ahol az idős nőbeteg is született. Felvetődött, hogy egy jelenleg 1600 fős faluból két, egymást követő generációban jelentkező, ritka betegség megjelenése nem lehet véletlen. A pontos családfa megrajzolása alapján kiderült, hogy a férfi beteg édesapjának elsőfokú unokatestvére az acromegalia miatt kezelt idős nőbeteg, ám korábban személyesen a két beteg nem is ismerte egymást (2., 3. ábra).

Megbeszélés
A hypophysis elülső lebeny daganatainak mintegy 5%-a előfordulhat öröklődő szindrómák részjelenségeként, mint az 1-es típusú multiplex endokrin neoplasia (MEN-1) és a Carney-komplex (CNC), illetve a familiáris izolált hypophysisadenoma (FIPA). Ezekben az esetekben a hypophysisadenoma kialakulásáért a MEN-1, a PRKAR1A, illetve az aril-hidrokarbon-receptorral interaktív protein (AIP) gén mutációja felelős (2). Az utóbbi években felismerésre került a MEN-1-hez hasonló szindróma vagy 4-es típusú multiplex endokrin neoplasia (MEN-4), amelynek hátterében a p27 fehérjét kódoló CDKN1B gén mutációja áll (3).
Következtetés
Egy családon belül előforduló hypophysistumorok kialakulása esetén beszélünk familiáris izolált hypophysisadenoma-, azaz FIPA-szindrómáról, amennyiben a MEN-1 és a Carney-komplex kizárható. A FIPA-családok 20-40%-ában mutatható ki az AIP gén mutációja, míg a családok nagyobb részénél a betegséget okozó gén még nem ismert. Az AIP-mutációt hordozó betegek esetében a tumor fiatalabb korban alakul ki, agresszív viselkedésű, nagyobb méretű és kevésbé reagál az SS-analóg kezelésre.
Tekintettel arra, hogy esetünkben a családban sem MEN-1, sem Carney-komplex nem fordult elő, és bár a hypophysisdaganat nem fiatal korban alakult ki, de mindkét beteg esetében nagyméretű volt és rosszul reagált SS-analóg-kezelésre, felmerült a FIPA, illetve akár az AIP-mutáció hordozásának lehetősége. Genetikai vizsgálat során AIP-mutációt (sem báziseltérést, sem pedig nagyobb géndeletiót) nem sikerült kimutatni, továbbá a MEN-1 és a CDKN1B gének mutációja sem volt igazolható.
Mivel a familiáris izolált hypophysisadenomában szenvedő családok nagyobb részénél a genetikai ok egyelőre nem ismert, így a potenciális hordozók esetében csupán biokémiai és képalkotó vizsgálatokat tudunk elvégezni, amelyek a férfi beteg gyermekei esetében negatívak lettek.
Irodalom
1. Daly AF, Rixhon M, Adam C. High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege, Belgium. J Clin Endocrinol Metab 2006;91:4769–4775.
2. Chahal HS, Chapple JP, Frohman LA, Grossman AB, Korbonits M. Clinical, genetic and molecular characterization of patients with familial isolated pituitary adenomas (FIPA). Trends Endocrinol Metab 2010;21(7):419–427.
3. Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci USA 2006;103(42):15558–15563.